
2020 - L’Azienda Agricola Sperimentale
L’acquisto dell’Azienda Agricola di Pugliano apre le porte alla produzione dei vini del futuro. Scopri di più
2020

Scorri
L’acquisto dell’Azienda Agricola di Pugliano apre le porte alla produzione dei vini del futuro. Scopri di più

Con la certificazione SQNPI La Guardiense è la prima cantina sostenibile in Campania Scopri di più

I Mille per l’Aglianico si aggiudica l’Oscar per la Miglior innovazione nel vino. Scopri di più
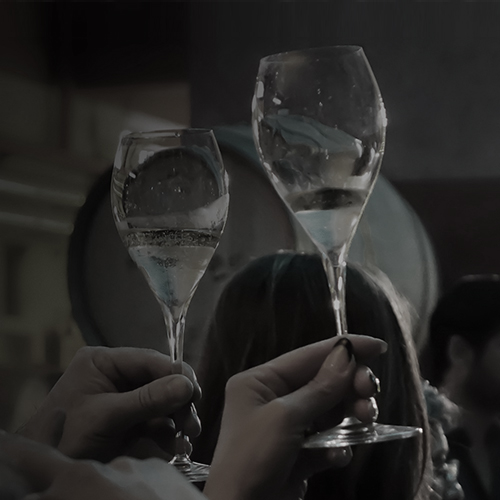
Si aprono le porte de La Guardiense, l’azienda diventa luogo d’incontro per la comunità e meta turistica campana. Scopri di più

L’enologo più conosciuto in Italia e all’estero accetta la sfida di collaborare con La Guardiense. Scopri di più

L’operato del presidente Domizio Pigna porta sostanziali cambiamenti alla rotta seguita fino a quel momento. Scopri di più

Si brinda al futuro con il primo spumante de La Guardiense che apre alla diversificazione dei prodotti. Scopri di più

I vini la Guardiense ricevono uno dei riconoscimenti più prestigiosi dell’epoca. La qualità dei vini sanniti comincia a farsi conoscere nel mondo. Scopri di più

Si vedono i primi frutti di tanto impegno, quelli che prima erano vignaioli ora diventano imprenditori a tutti gli effetti Scopri di più

La determinazione incrollabile dei trentatré soci riceve la fiducia delle istituzioni. Scopri di più

Lungimiranza e coraggio animano lo sforzo di 33 soci che si impegnano per rivoluzionare la viticoltura nel Sannio e non solo. Scopri di più